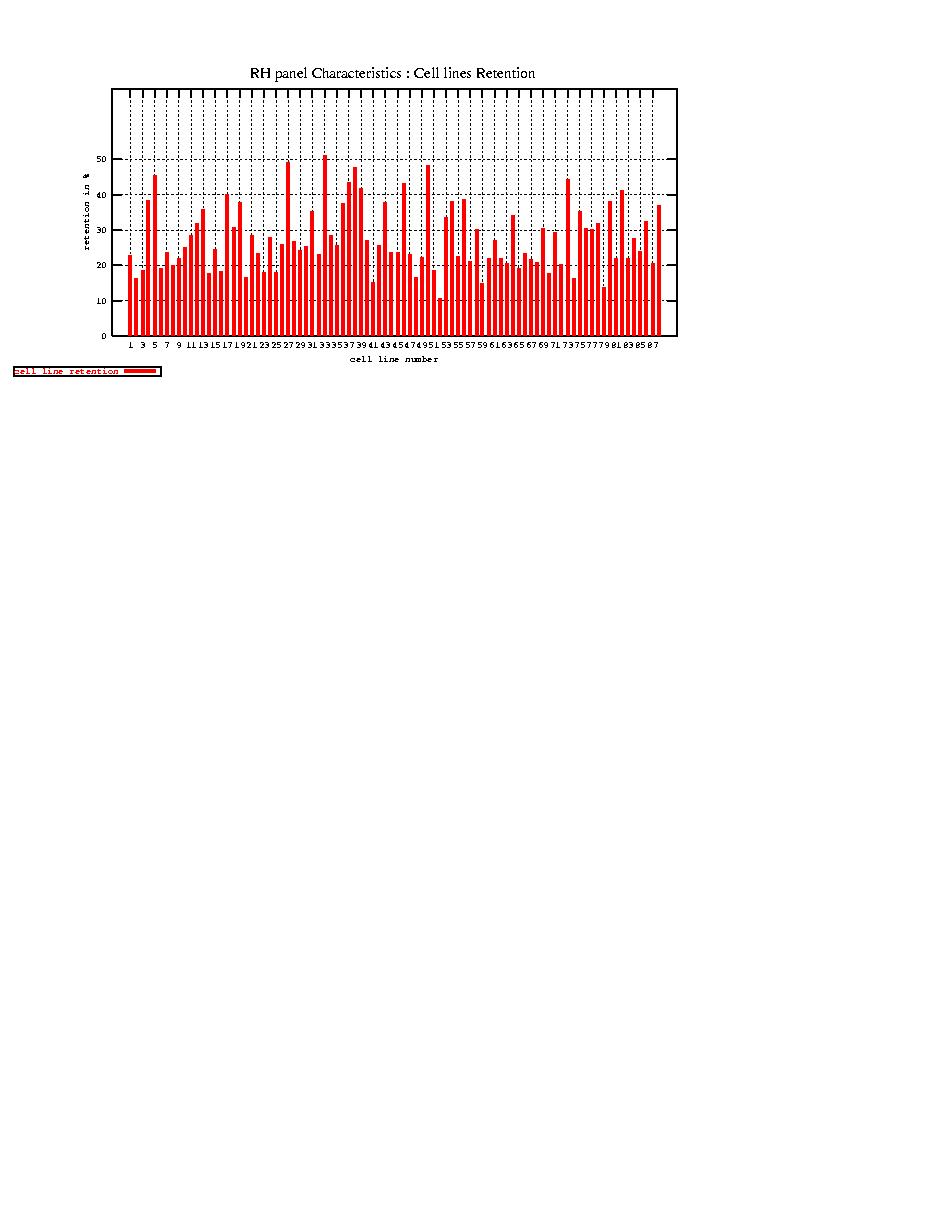
- Cell lines retention
(percentage are derived from 10,500 RH genotyping data).
Cell Line n.1 = 23.0071 %
Cell Line n.2 = 16.3397 %
Cell Line n.3 = 18.6298 %
Cell Line n.4 = 38.3902 %
Cell Line n.5 = 45.3957 %
Cell Line n.6 = 19.1613 %
Cell Line n.7 = 23.8477 %
Cell Line n.8 = 20.0019 %
Cell Line n.9 = 22.0118 %
Cell Line n.10 = 25.2005 %
Cell Line n.11 = 28.4858 %
Cell Line n.12 = 32.0224 %
Cell Line n.13 = 35.7909 %
Cell Line n.14 = 17.7215 %
Cell Line n.15 = 24.5531 %
Cell Line n.16 = 18.4559 %
Cell Line n.17 = 40.0425 %
Cell Line n.18 = 30.7952 %
Cell Line n.19 = 37.8104 %
Cell Line n.20 = 16.7552 %
Cell Line n.21 = 28.4279 %
Cell Line n.22 = 23.5578 %
Cell Line n.23 = 18.1177 %
Cell Line n.24 = 27.8771 %
Cell Line n.25 = 18.0501 %
Cell Line n.26 = 26.0798 %
Cell Line n.27 = 49.3091 %
Cell Line n.28 = 26.7949 %
Cell Line n.29 = 24.2246 %
Cell Line n.30 = 25.4517 %
Cell Line n.31 = 35.4334 %
Cell Line n.32 = 23.2969 %
Cell Line n.33 = 51.0387 %
Cell Line n.34 = 28.5148 %
Cell Line n.35 = 25.674 %
Cell Line n.36 = 37.5882 %
Cell Line n.37 = 43.5791 %
Cell Line n.38 = 47.8404 %
Cell Line n.39 = 41.8688 %
Cell Line n.40 = 27.0268 %
Cell Line n.41 = 15.3445 %
Cell Line n.42 = 25.7996 %
Cell Line n.43 = 37.965 %
Cell Line n.44 = 23.5965 %
Cell Line n.45 = 23.7318 %
Cell Line n.46 = 43.2022 %
Cell Line n.47 = 23.21 %
Cell Line n.48 = 16.533 %
Cell Line n.49 = 22.2534 %
Cell Line n.50 = 48.2655 %
Cell Line n.51 = 18.5718 %
Cell Line n.52 = 10.5904 %
Cell Line n.53 = 33.5395 %
Cell Line n.54 = 38.1486 %
Cell Line n.55 = 22.5722 %
Cell Line n.56 = 38.738 %
Cell Line n.57 = 21.1711 %
Cell Line n.58 = 30.1092 %
Cell Line n.59 = 15.0449 %
Cell Line n.60 = 21.9152 %
Cell Line n.61 = 27.1041 %
Cell Line n.62 = 22.0118 %
Cell Line n.63 = 20.6687 %
Cell Line n.64 = 34.0613 %
Cell Line n.65 = 19.1323 %
Cell Line n.66 = 23.3936 %
Cell Line n.67 = 21.7992 %
Cell Line n.68 = 20.8909 %
Cell Line n.69 = 30.5827 %
Cell Line n.70 = 17.7215 %
Cell Line n.71 = 29.3458 %
Cell Line n.72 = 20.3015 %
Cell Line n.73 = 44.4487 %
Cell Line n.74 = 16.2335 %
Cell Line n.75 = 35.2401 %
Cell Line n.76 = 30.3701 %
Cell Line n.77 = 30.1962 %
Cell Line n.78 = 32.0128 %
Cell Line n.79 = 13.9434 %
Cell Line n.80 = 38.2452 %
Cell Line n.81 = 21.9152 %
Cell Line n.82 = 41.2407 %
Cell Line n.83 = 22.1084 %
Cell Line n.84 = 27.7708 %
Cell Line n.85 = 23.9443 %
Cell Line n.86 = 32.3799 %
Cell Line n.87 = 20.6977 %
Cell Line n.88 = 37.0374 %
RH panel average retention : 29 %
|
Histogram of cell line retention [-full size-]
 |